

TL;DR we compared a bunch of protein language models in the lab. While there are winners (see below!), how you use the model matters about as much as which model you choose, and having even a small amount of data is far better than any zero-shot method.
Protein Language Models and Antibody Drug Engineering
Generative protein language models (pLMs) have become a core part of the modern AI/ML-driven protein engineering workflow, and so at BigHat, these are one of the many tools we deploy in our weekly design-build-test loop. These models have the same architecture and pre-training as the LLMs we have all come to know/love/hate, but they learn to ‘speak’ the language of amino acids.
There has been a proliferation of these models in recent years, with some focusing on antibody-specific training data, some playing the same scaling game that has been so successful in natural-language LLMs, and others tweaking the training process and objectives. Because we’ve often seen a difference between the metrics reported in publications and real-world performance, and because it’s increasingly clear that bigger isn’t always better with pLMs1, we decided to perform an evaluation of our own.
The Bakeoff
Our goal was to find the model that was best at diversifying existing antibody sequences, without ‘breaking’ the molecule; we want to be able to introduce multiple mutations in the CDRs (the region responsible for binding the target) and still have the resulting variants be producible in the lab and retain binding. This sort of task is useful for ‘hit expansion’ - turning one hit into many - as well as for initial exploration and dataset generation at the beginning of an optimization campaign.
We surveyed the literature, focusing on sequence-only, open-source pLMs, and identified >30 models of interest. From these, we identified 11 models that covered a range of important properties - antibody-specific or general, autoregressive or masked language model, large or small, etc. These models underwent an initial in silico evaluation, including some baseline models for comparison (one structure model and one lightweight model). We used these models to propose mutations for leads from an optimization campaign, and then used a suite of in-house predictive models to evaluate the resulting models across 6 metrics (Figure 1). We also tested different methods for generating variants with the same models (compare e.g., Gibbs and Beam Search - more on this later!).
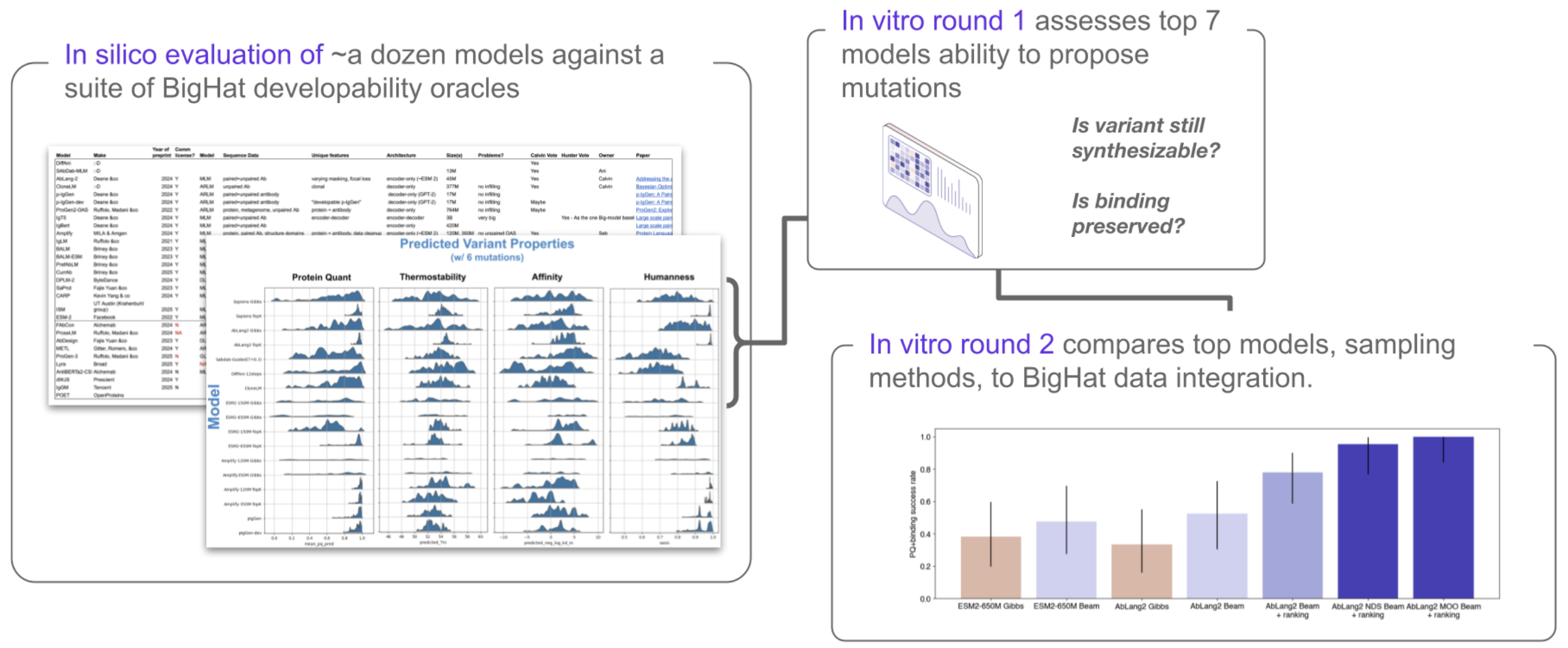
From this, we selected 7 models to send to the lab, designing ≥20 variants of both scFvs and Fabs for each (along with structure and lightweight model baselines). The best-performing models from the first round of in vitro testing were included in a second round, and the final results are interesting from several perspectives (Figure 2). It’s worth noting that an evaluation like this, including many models and many plates of variants, is an easy, routine task rather than a chore thanks to RADS.

What did we learn?
Ablang2 has a narrow lead over other models, though the differences are quite small despite the years of pLM development and dozens (hundreds?) of publications.
Sampling method matters as much or more than the choice of model - every model is better with beam search, and the per-model improvement from sampling method is bigger than the delta between most models. See our publication “How to make the most of your masked language model for protein engineering” for more details on sampling methods.
Models vs. Data
So far, all the results we’ve discussed could be described as “zero-shot”: the model is pre-trained on a large public dataset, and then we apply it to sequences from a particular drug development campaign that certainly differ from the public training data. However, at BigHat, campaign-specific data generation is our superpower, so it makes sense to ask how helpful this data might be. Given how expensive training large pLMs can be, this may even be the cheaper option!
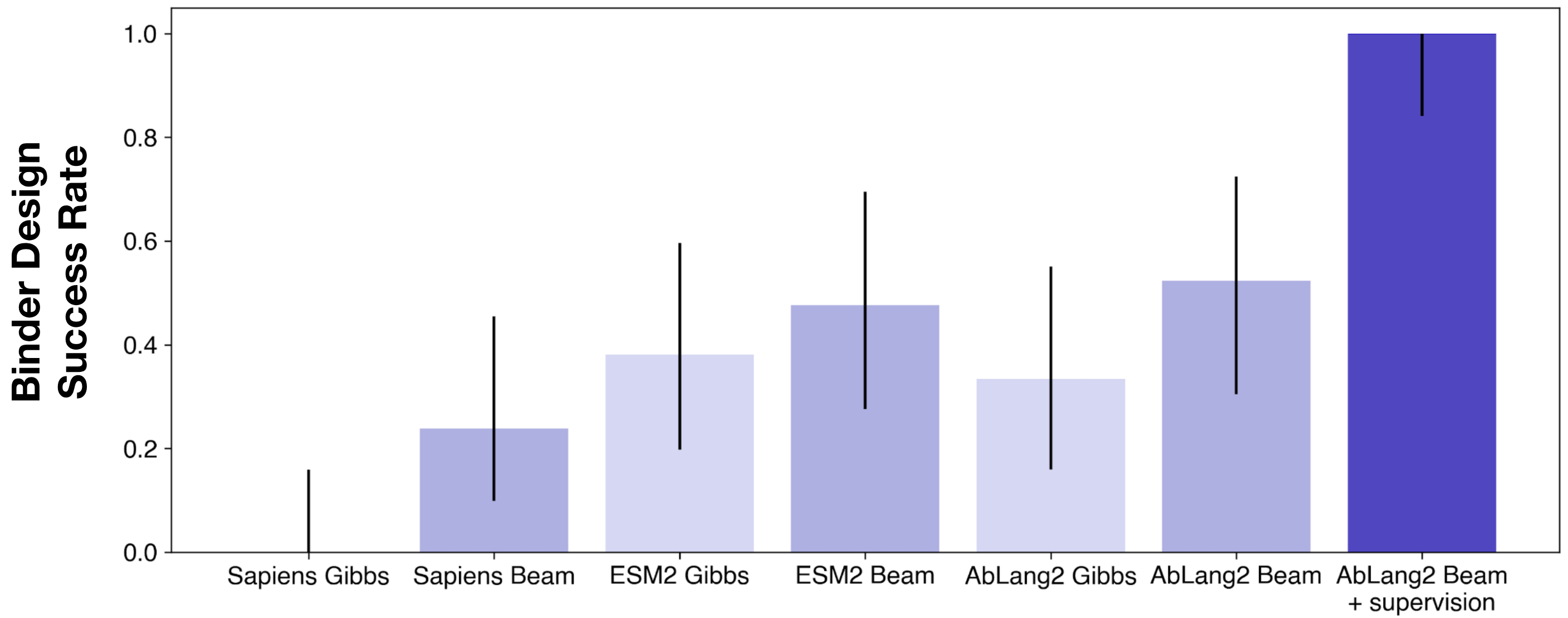
We find that using even a small amount of data (~400-700 samples2, less than a week of BigHat platform capacity) to guide the generative model and filter its outputs yields by far the best performing model, and provides a performance improvement bigger than the past few years of pLM model development (Figure 3. See “How to make the most of your masked language model for protein engineering” for more details).
So what?
There are a few high-level points worth taking away from this (though see below for some more in-the-weeds but interesting findings!):
- Choice of model does matter somewhat, and Ablang2 is a great starting point for CDR diversification.
- How you use the model matters as much or more than which model you use! Try beam search with your favorite pLM for a free performance boost.
- If you can collect data, do it! Incorporating data from your drug development program is by far your best option and may save you time and money compared to the GPU hours you might burn pre-training a better pLM.
Aside: Structure vs. Sequence
We also evaluated a model (DiffAb+) that includes structural information for comparison to sequence-only models. The overall success rate for this model was modest, but perhaps more interestingly, the variants for which it was successful seemed to be biased towards tighter binding. Is this because of the structural information, or could it be simply a difference in the training data, because this model is trained on the PDB instead of e.g., OAS? Comparing the results to a pLM trained only on the sequences from the PDB (no structures) provides some insight: at least some of the improvement in binding comes solely from the bias in the sequences contained in the PDB! (Figure A1). While only a tantalizing hint, this adds to a growing body of evidence that the biases in what people bother to determine structures for can also result in biases in models.

For a more in-depth look at our work, please see our recent publication:
References




